
Application areas
Simulations are created in our institute in a wide variety of areas. Whether the hydration process of cement, the heat flow through different materials, transport mechanisms or the interaction of concrete with other materials are to be investigated, each question can not only be practically demonstrated in our laboratories but also supported by simulations.
These questions can be considered in the individual direction by themselves or in combination.
Not only different influences can be combined, but also different scales. For example, a multiscale model can provide the influence of microstructure on meso- or macroscopic material properties.
Applications
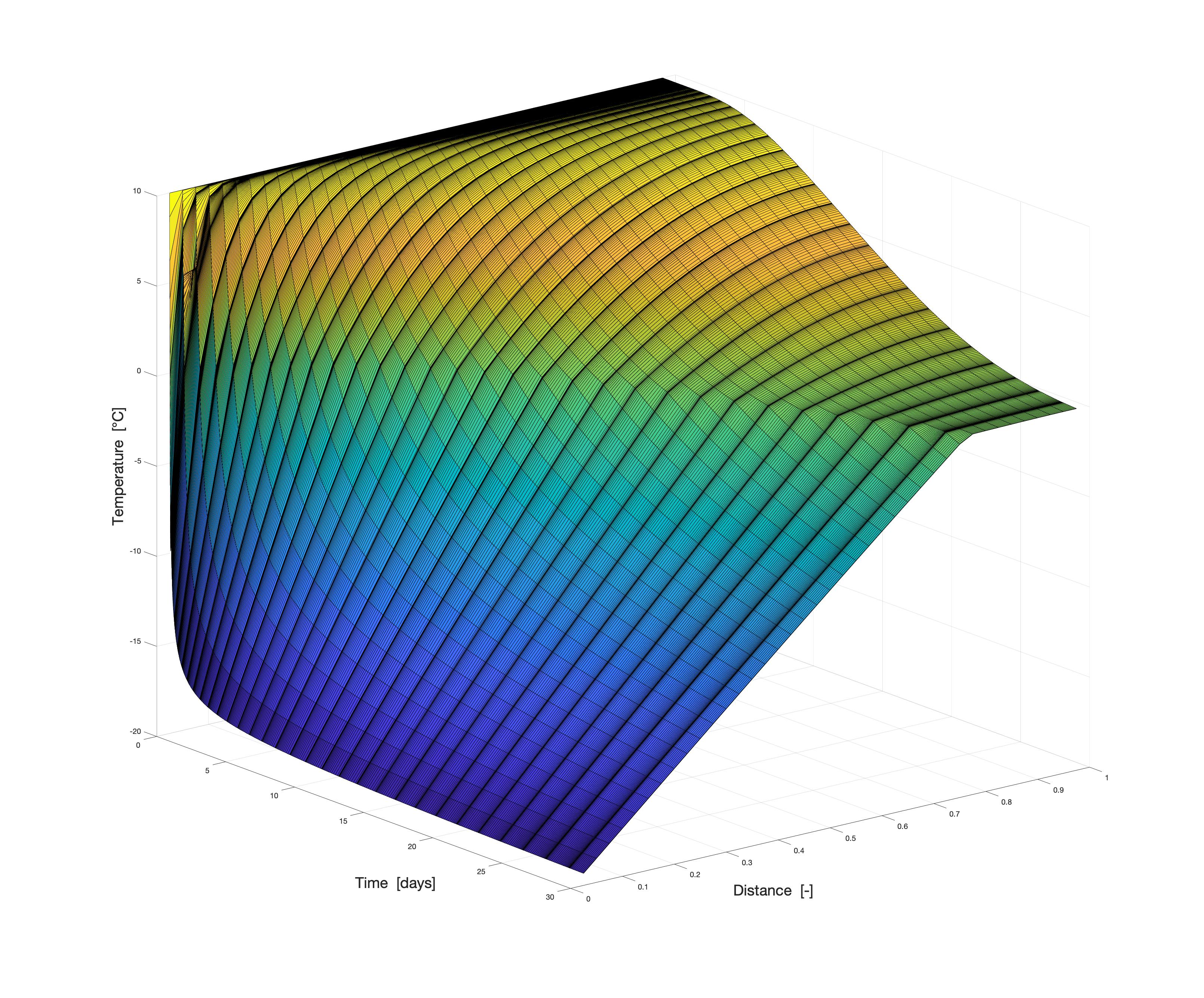
In order to be able to cover the broad spectrum, programs or development environments that can adapt to the challenges are also necessary. Therefore, we also work with software solutions tailored to the issues.
For example, commercial software such as Abaqus can be used for direct proofs and verifications. Open source software, on the other hand, can offer greater freedom in research.
However, environments such as Matlab or Python also help with evaluation and research.
For looking at multiple variable influences or applying the phase field method, multiphysics solutions such as MOOSE Framework or openFoam are also used.
Experiences in various numerical applications:
Some applications that have already been used, in some cases in different areas, include:
- Abaqus
- Hymostruc
- MOOSE Framework
- OpenFOAM
- PHREEQC
- LAMMPS Molecular Dynamics Simulator
- Matlab
This list is continuously expanded to achieve the best solutions for our research and for investigations.
Computational resources
Depending on the problem, different computing power is required.
While the computing power of the standard computers available on the market may be sufficient for static, linear calculations, increased resolutions or nonlinearities may already exceed these resources.
To be prepared for this, capacities are available at different levels:
In addition to the normal workstation computers, computers optimized for numerical simulations are also available.
These are computers with:
- 8-core processors
- Up to 128GB RAM for non-linear calculations with a high number of degrees of freedom
- NVIDIA graphics cards for GPU acceleration with Cuda
High Performance Computing at WiB

Our Institute own a High Performance Computer (HPC). The system is build with the power of a dual socket Supermicro GPU node with AMD EPYC™ Milan CPUs and 512GB of memory. This state-of-the-art technology will enable us to perform complex computations at high speeds and accuracy.
The AMD EPYC™ processors with a high core count are designed specifically for HPC workloads, delivering superior performance, scalability, and energy efficiency.
Additional graphic cards (GPUs) will accelerate compute-intensive tasks such as machine learning, deep learning, and scientific simulations. The combination of these powerful CPUs and GPUs enables faster data processing, leading to quicker time-to-insight and better decision making.
Furthermore, both nodes comes with together 768GB of DDR4 RAM, allowing for efficient handling of massive amounts of data as well as the computation of large systems with a high amount of nodes. This also ensures that even the most demanding applications run smoothly without experiencing any bottlenecks or slowdowns.
With this advanced hardware configuration, our HPC system is capable of performing a wide range of calculations, enabling our team to tackle some of the most challenging problems.
These capabilities allowing us simulating
- complex systems such as heterogenous materials with millions of nodes
- nonlinear materials behavior where high precision is needed
- multi-physics simulations involving the complex coupling of its own set of PDEs (partial differential equations) utilizing the parallel processing capabilities of the CPU and GPU architectures
- analyzing vast amounts of data, or developing cutting-edge AI models
- etc.
This leads to more detailed and realistic simulations of complex systems.

High Performance Computing
If the computing power is not sufficient, we already have experience with the Lichtenberg high-performance computer. Here, for example, parallel calculations with a large number of processors can be used by means of Message Passing Interface (MPI), so that even very complex problems can be solved.
This means that the computing power at the WiB is not a limiting factor.
Lichtenberg high-performance computer at the TU Darmstadt
The Lichtenberg High Performance Computer, part of the Hessian Competence Center for High Performance Computing, provides users with large computing capacities of up to 3.148 PFlop/s and 257 TBytes of RAM.
More info can be found here:
Presentation of some research areas
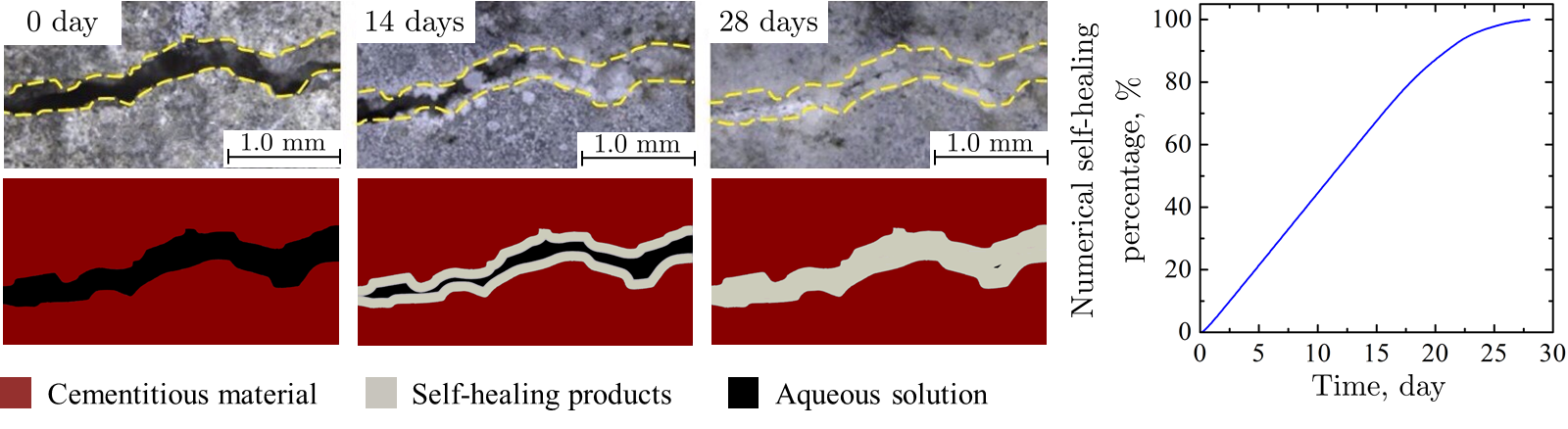
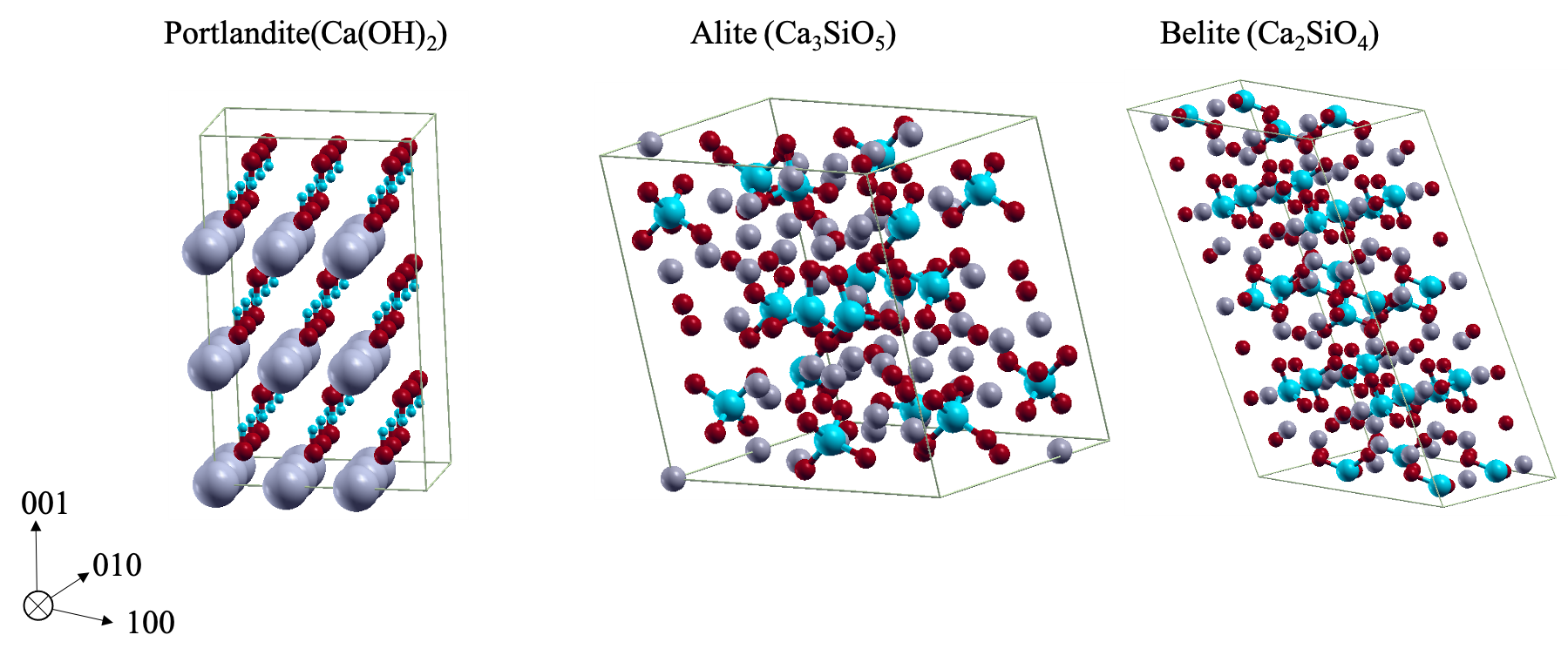

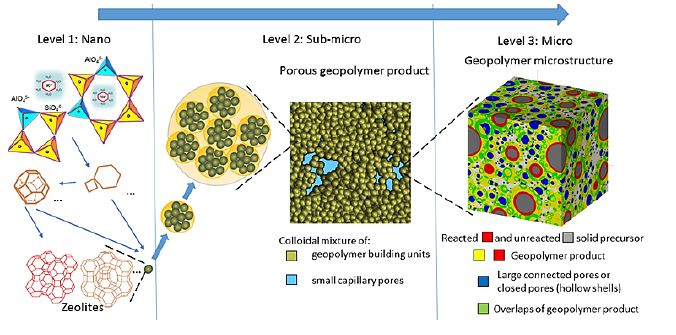
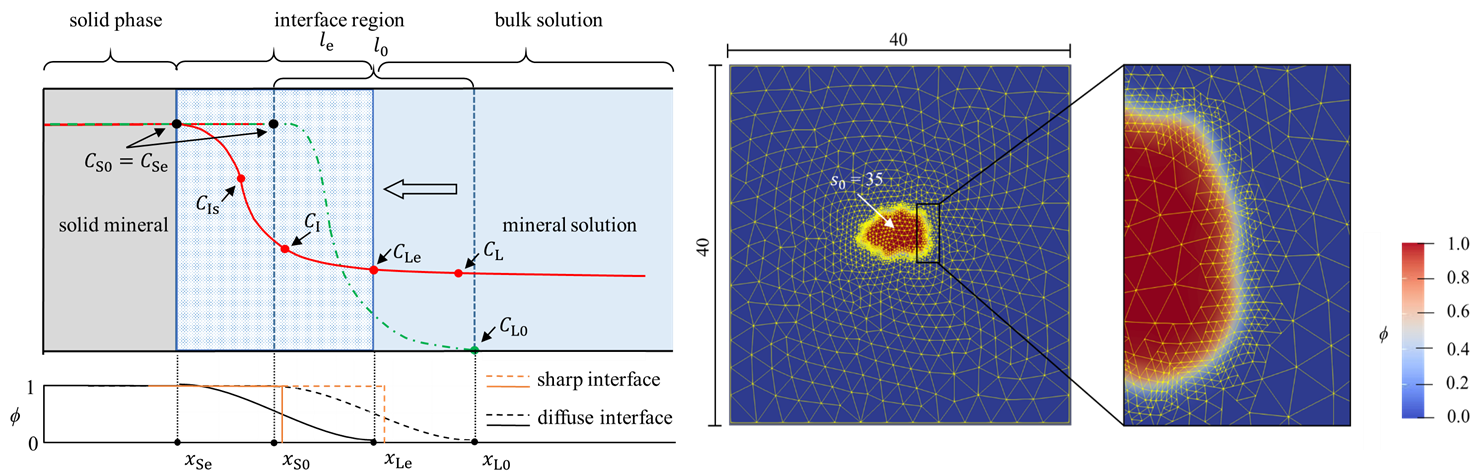
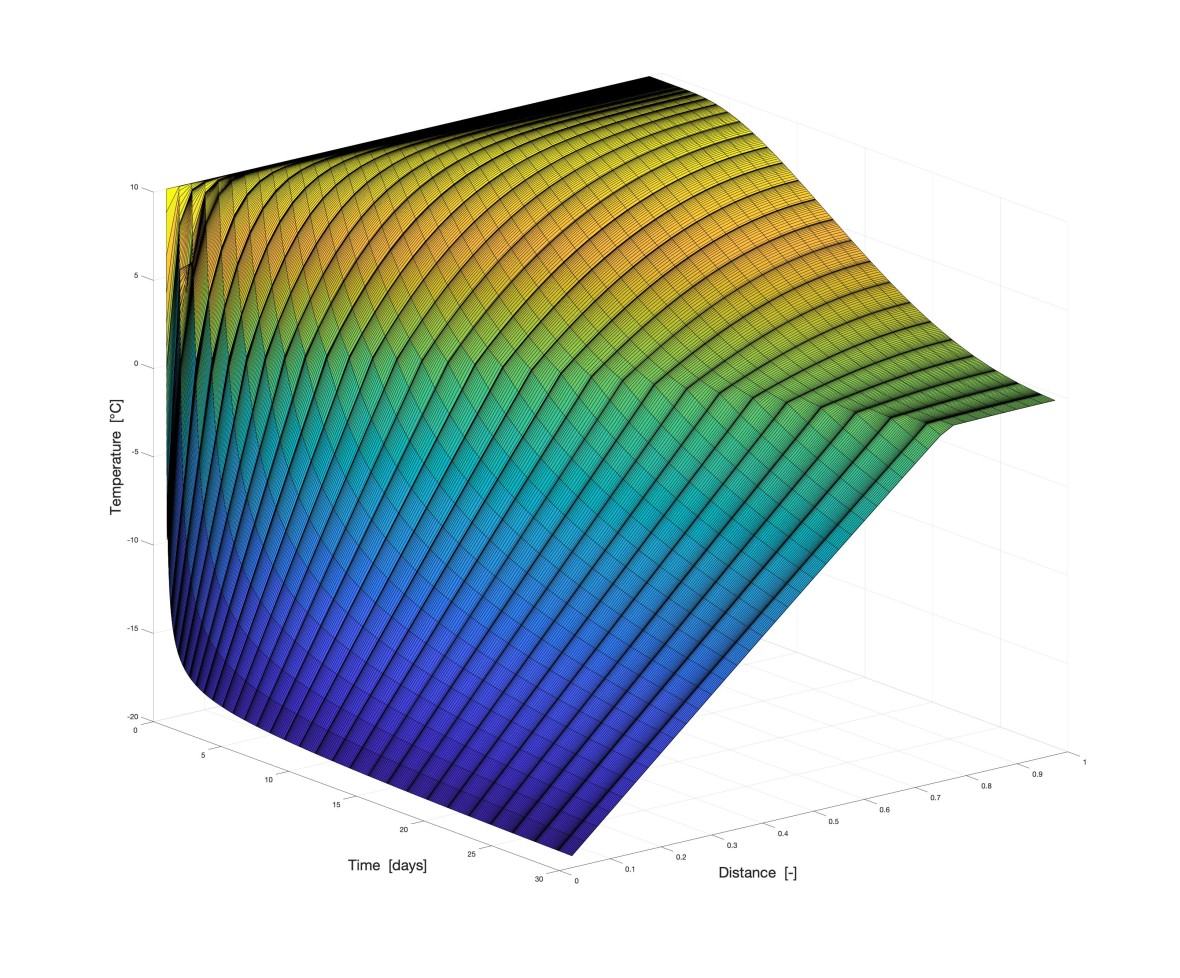
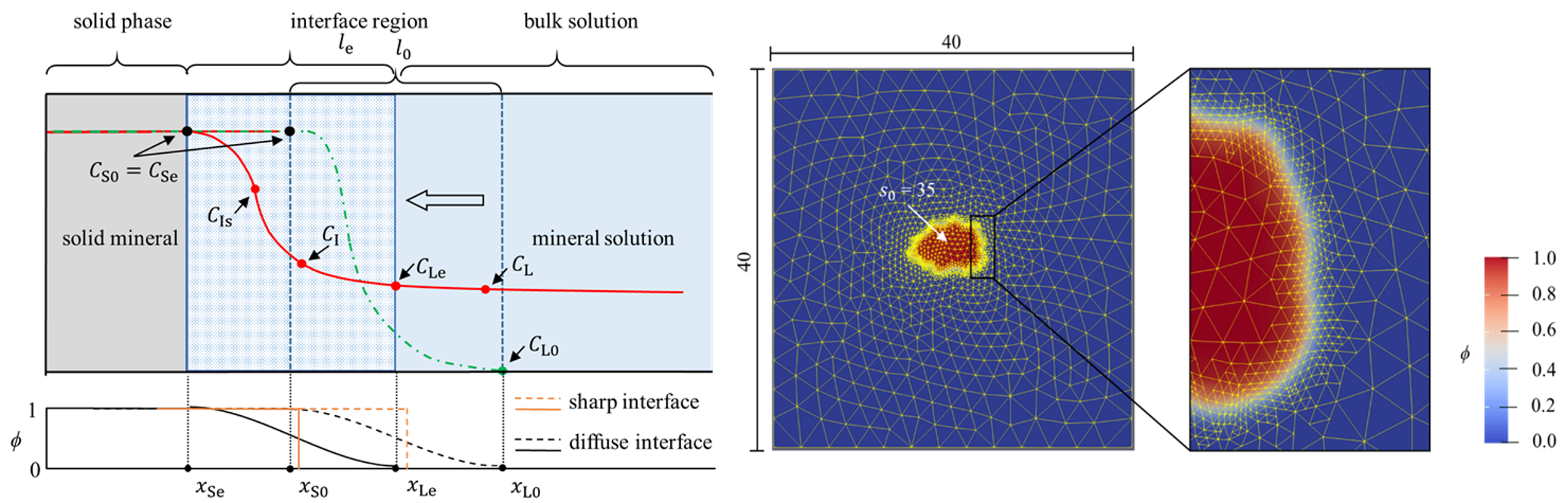
Simulations done at the WiB
Here you can see some examples of past and current simulations at our institute:





- Go to picture 1
- Go to picture 2
- Go to picture 3
- Go to picture 4
- Go to picture 5
- Go to picture 6
- Go to picture 7