Project abstract
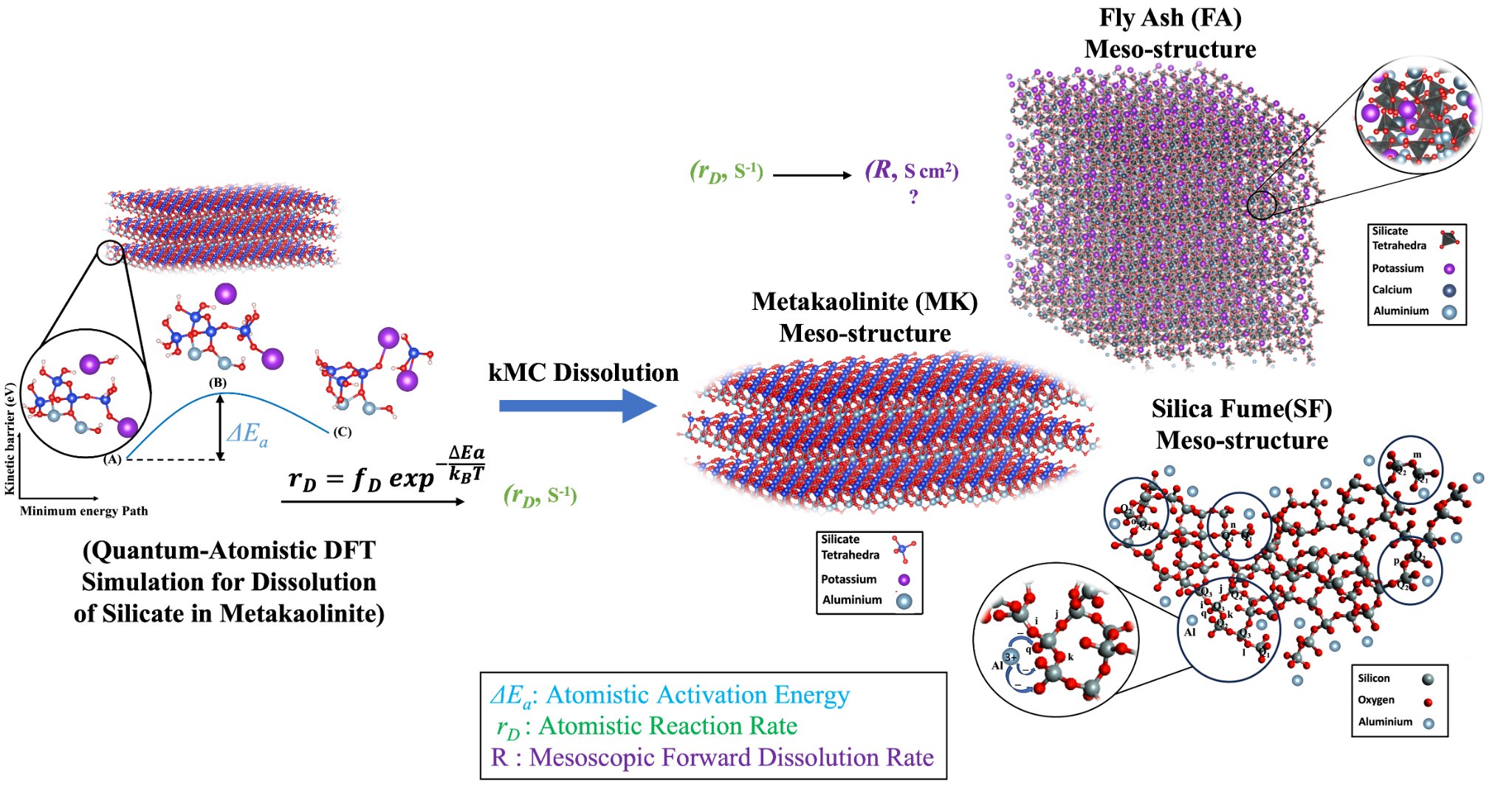
There is a strong demand to link upscaling atomistic dissolution mechanisms with the mesoscopic pozzolans dissolution reactivity. This could be achieved only after being able to predict (initial) forward dissolution rate constants (k+) for each crystal surfaces. Atomistic modelling approaches are needed to compute the reaction rates for dissolution of a) Si(O)44- (silica fume) vs. SiO44-Al3+ (fly ash, slags) b) meta(dis)kaolinite crystalline clay structures: Si(O)44- vs. Al., initially far-from-equilibrium to obtain a fundamental (material’s structural) parameter decoupled from solution (Si and Al) concentrations and precipitation reactions. According to the different distributions of individual crystal in mesoscopic pozzolans and meta-clay structures by employing the KMC upscaling approach, the missing connection between atomistic, and meso-structures for solving the net forward dissolution rate constant can properly be solved. In this way, it is needed to use the quantum chemistry DFT (in VASP software) using density-functional-perturbation theory (DFPT) for the calculation of vibrational frequencies V (s-1) at the reactant and saddle point. Moreover, the enthalpy activation energy (Ea) can be computed for forward dissolution of ions at far-from-equilibrium condition, using the improved dimer method (IDM) and the transition-state theory (TST) within the density functional theory.
Applicants: Izadifar, TUDa